The emergence of PROTACs has changed the mindset of researchers targeting disease-causing proteins. By shifting the approach from traditional inhibition to targeted degradation, this new modality offers a novel solution for handling proteins that were previously considered “undruggable,” using the cell’s natural protein degradation machinery (2) .
Every innovative leap forward adds extra layers of complexity, and PROTACs are no exception. As the pharmaceutical industry’s interest in these molecules grows, early-stage development, especially the path from lead optimization to Investigational New Drug (IND) submission, has emerged as a critical challenge in advancing more PROTAC candidates into clinical development (3,4).
However, by examining the strategies and lessons learned from early-stage PROTAC development, sponsors can bring more PROTACs closer to patients.
The Unique Structure and Mechanism of PROTACs
PROTACs are heterobifunctional small molecules that leverage the body’s natural ubiquitin-proteasome system to degrade target proteins selectively. Unlike traditional inhibitors that block protein functions through direct binding, PROTACs work as catalysts by bringing together a target protein and an E3 ubiquitin ligase, resulting in ubiquitination and subsequent proteasomal degradation of the target protein (5).
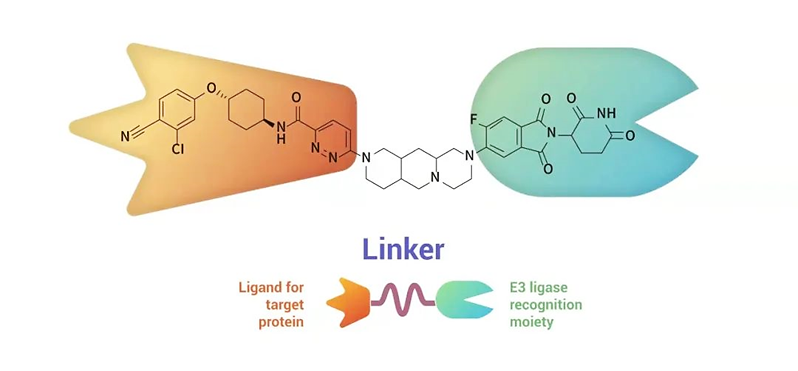
A PROTAC molecule contains three elements, as shown in Fig. 1:
• A ligand that specifically binds to the target protein,
• A ligand that specifically binds to an E3 ubiquitin ligase (e.g., VHL, CRBN), and
• A linker that connects them.
After administration, a PROTAC molecule brings a target protein into proximity with an E3 ubiquitin ligase, initiating the transfer of ubiquitin onto the target. Once tagged with ubiquitin, the protein is recognized and degraded by the proteasome in the cells. Because the molecule behaves as a catalyst, a single PROTAC can trigger the degradation of multiple copies of the target protein. This approach offers a powerful means of eliminating disease-driving proteins, including those that lack enzymatic activity or are otherwise considered difficult to target with conventional drug design.
Figure 1. Schematic of a typical PROTAC structure and an example.
Drugability of PROTAC Molecules
PROTAC molecules typically have a molecular weight range of 800 to 1000 Daltons and contain many polar groups, as well as hydrogen bond donors and acceptors. PROTACs frequently break Lipinski’s Rule of Five, which is regarded as a general criterion for good oral drug-likeness. As a result, PROTACs may suffer from problems such as reduced oral bioavailability, poor membrane permeability, and restricted metabolic stability.
A combination of medicinal chemistry and formulation is needed in the early development process to address these concerns. Researchers must optimize linker length, minimize the polar surface area, and simplify the ligand structure wherever possible (6,7). Regarding formulation, technologies such as amorphous solid dispersions, lipid-based delivery systems, and prodrug approaches can enhance in vivo exposure (8).
Improving the oral drug-likeness of PROTACs is a critical goal in drug discovery efforts. Developing molecules suitable for oral dosage adds convenience for patients and supports continuous treatment regimens.
Metabolism and Stability Challenge: Early Soft Spot Identification
Due to their chemical design and structure, PROTACs are susceptible to metabolic degradation, especially at linker regions. According to data from systematic metabolite identification studies, more than 70% of PROTACs undergo fast biotransformation at or near the aliphatic linker, most often via CYP3A-mediated oxidation followed by hydrolysis or further oxidative cleavage (9).
Conventional in vitro assays, such as those using liver microsomes, may not be sufficient to understand the metabolism and stability of PROTACs, as they lack the complete enzymatic complement responsible for both phase I and phase II transformations (10).
A more comprehensive in vitro approach should be used to improve the early detection of the metabolic soft spots and to guide structure optimization, including:
• Hepatocytes: Provide a full range of metabolic enzymes and better represent in vivo metabolism.
• Liver S9 fractions: Enable detection of both oxidative and conjugative metabolism.
• Cytosolic fractions: Allow screening for non-CYP enzymes such as aldehyde oxidase (AO), which may contribute to rapid clearance and demonstrate species-specific activity.
PROTAC developers can proactively detect and address metabolic soft spots by integrating these techniques early in the preclinical stage. This approach minimizes the risk of late-stage failures and boosts confidence as programs advance into toxicology and first-in-human studies.
Analytical Characterization Challenges
Bioanalysts often approach PROTACs as traditional small molecules, and their analytical characterization during early development presents many unique challenges (11). Due to their large molecular weight, structural complexity, and the presence of multiple stereocenters, PROTACs often exhibit analytical artifacts, including ion suppression, peak splitting during chromatographic separation, and in-source fragmentation, in LC-MS/MS assays. PROTACs also tend to show high non-specific binding to plasticware and glassware in the lab, compromising accurate quantitation and assay reproducibility.
To overcome these obstacles, developers must use low-binding bioanalytical techniques. Careful optimization of mass spectrometry parameters, along with the use of desorbents such as Tween 20, can significantly improve signal stability and data quality (11). Additionally, plasma stability should be evaluated in fresh and frozen matrices, as changes in enzyme activity and protein integrity may impact the interpretation of stability findings.
Pharmacokinetics and the PK/PD Relationship
Understanding the pharmacokinetic (PK) and pharmacodynamic (PD) characteristics of PROTACs poses distinct challenges that are not typically faced by traditional small molecules. Since PROTACs behave catalytically, the relationship between systemic exposure and biological effect is often non-linear. Complexity is increased by the “hook effect”, in which high concentrations of PROTACs reduce activity due to saturation of binary complexes (12). Addressing these problems requires a shift in approach:
• Mechanistic PK/PD models can better account for catalytic and time-dependent effects.
• Efficacy correlates more closely with degradation kinetics (e.g., maximum degradation, degradation EC50, and degradation half-life) than steady-state concentrations.
• Dosing regimens need careful optimization to maintain efficacy without causing saturation or loss of selectivity.
Cellular Uptake and Intracellular Localization
Despite sufficient extracellular exposure, many PROTACs fall short intracellularly due to poor membrane permeability and large polar surface areas. This can severely limit ternary complex formation and reduce degradation efficiency, especially in hard-to-penetrate tissues or tumor microenvironments.
Several strategies can improve cellular uptake. Optimizing physicochemical attributes such as molecular weight, lipophilicity, polarity, and conformational flexibility can help. Utilizing nanoparticle-based or lipid-assisted delivery systems to overcome cellular barriers can enable active targeting or membrane translocation.
Developers should also leverage prodrug strategies such as masking polar functional groups with cleavable moieties to enhance membrane permeability and enable intracellular activation through enzymatic cleavage (13).
Selection of E3 Ligases
Most PROTACs rely on a few well-characterized E3 ligases, such as VHL and CRBN. However, these may not offer tissue-specific degradation due to the ubiquitous expression of E3 ligases. This could result in unwanted degradation in non-target tissues. The industry is now investing in discovering and validating novel E3 ligases with restricted tissue expression to minimize off-target effects and improve therapeutic windows.
The next frontier of innovation is already underway and includes efforts to discover E3 ligases with tissue or disease-specific expression patterns. Structure-based screening is used to identify new ligands and chemotypes for less-characterized E3s. And ternary complex dynamics, including cooperativity and off-rates, are incorporated into ligand selection to optimize potency and selectivity.
On-Target and Off-Target Toxicity Effects
Clear safety risks are associated with the therapeutic potency of PROTACs, which is crucial to their ability to degrade target proteins completely. Unlike inhibitors that modulate function temporarily, PROTACs can lead to irreversible consequences if the target plays a role in essential cellular functions. Unintended toxicity can arise even from the low-level degradation of essential or partially homologous proteins, causing on-target toxicity.
Furthermore, E3 ligases carry some risk: their recruitment may lead to off-target ubiquitination and degradation, especially if they are highly expressed or broadly active across tissues (14). This is especially important when considering overexpressed ligases, such as CRBN. Additionally, linker chemistry and metabolic byproducts can contribute to toxicity by forming reactive or unstable intermediates.
Several strategies have been explored to reduce both on-target and off-target toxicities associated with PROTACs during the preclinical stage.
• Global proteomic profiling is increasingly applied to map the degradation profile across various cell types and species, making it possible for early identification of unintended protein targets (15).
• Genetic screens can assess gene essentiality in disease-relevant and normal tissues, providing useful information and insights on possible on-target liabilities.
• Incorporating delivery strategies, such as nanoparticles, antibody-PROTAC conjugates, or prodrug formulations, can help modulate tissue distribution and reduce exposure to non-target organs, especially when systemic degradation poses a safety risk.
• Metabolite identification studies, particularly in vivo, are crucial for detecting toxic degradation products or reactive linker-derived fragments.
• Tissue cross-reactivity and biodistribution studies are essential for evaluating unintended effects in non-target organs, especially when E3 ligases or target proteins are expressed outside of the intended therapeutic window.
Utilizing the Latest Technology for Efficiency
As PROTAC discovery becomes increasingly time-consuming and resource-intensive, computational and machine learning tools are playing a growing role in accelerating early-stage development (16,17). These technologies are now being utilized to predict ternary complex formation, assess degradation potential, and inform the design of optimal linkers and ligands prior to compound synthesis.
By integrating diverse data sources, from structural models to cellular degradation outcomes, computational platforms can help prioritize candidates with the highest likelihood of success. These approaches are still evolving but are already reducing reliance on broad empirical screening and enabling a more focused, data-driven path to selecting effective and developable PROTACs.
Integration and Cross-Functional Planning
A successful IND submission for a PROTAC program ultimately depends on the seamless integration of chemical optimization, ADME characterization, analytical method development, and toxicology. Best practices now emphasize early and frequent cross-functional collaboration to ensure that degradation efficacy is not pursued at the expense of bioavailability or safety. Cross-functional collaboration, established early and maintained throughout, is proving essential to navigating the complex path to the clinical stages. Best practices emerging from successful programs include:
• Early alignment on developability thresholds, including synthetic accessibility, stability, permeability, and formulation feasibility.
• Real-time integration of bioanalytical, DMPK, and efficacy data into decision-making frameworks.
• Adoption of scoring or ranking systems to compare candidates holistically, not just on degradation potency.
A Final Word
The NDA submission of vepdegestrant marked more than just a regulatory milestone for PROTACs as a new therapeutic modality. The promise of PROTACs is beginning to translate from laboratory research to patient benefit. However, as the first few PROTACs move from concept to clinic, the path forward remains far from straightforward. Early-stage PROTAC development presents unique scientific challenges, including optimizing chemical structures and metabolic stability and managing PK/PD dynamics, bioanalysis, metabolism, safety, and other factors.
As more programs follow in the footsteps of early pioneers, success will hinge on recognizing that PROTACs are not simply modified small molecules—they are a distinct modality that requires their own development playbook. By integrating cross-functional insights early, embracing data-driven and mechanistic strategies, and staying ahead of emerging risks, PROTAC drug developers can better navigate the path to IND and ultimately to NDA.
Author
 Shanghao Li, Ph.D., International Marketing Associate Director in the Laboratory Testing Division at WuXi AppTec, has expertise in protein characterization, novel biomaterials, targeted drug delivery, and bioanalytical method development, supporting integrated DMPK, Bioanalysis, and Toxicology solutions.
Shanghao Li, Ph.D., International Marketing Associate Director in the Laboratory Testing Division at WuXi AppTec, has expertise in protein characterization, novel biomaterials, targeted drug delivery, and bioanalytical method development, supporting integrated DMPK, Bioanalysis, and Toxicology solutions.
References
1. Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. Protacs: Chimeric molecules that target proteins to the Skp1–Cullin–F box complex for ubiquitination and degradation. Proceedings of the National Academy of Sciences. 2001;98(15):8554-9.
2. Kim J, Park CB. Shedding light on biocatalysis: photoelectrochemical platforms for solar-driven biotransformation. Current Opinion in Chemical Biology. 2019;49:122-9.
3. Guedeney N, Cornu M, Schwalen F, Kieffer C, Voisin-Chiret AS. PROTAC technology: A new drug design for chemical biology with many challenges in drug discovery. Drug Discovery Today. 2023;28(1):103395.
4. Zeng S, Huang W, Zheng X, Zhang Z, Wang J, Shen Z. Proteolysis targeting chimera (PROTAC) in drug discovery paradigm: Recent progress and future challenges. European Journal of Medicinal Chemistry. 2021;210:112981.
5. Sakamoto KM, Kim KB, Verma R, Ransick A, Stein B, Crews CM, et al. Development of Protacs to target cancer-promoting proteins for ubiquitination and degradation. Molecular & Cellular Proteomics. 2003;2(12):1350-8.
6. Troup RI, Fallan C, Baud MG. Current strategies for the design of PROTAC linkers: a critical review. Exploration of Targeted Anti-tumor Therapy. 2020;1(5):273.
7. García Jiménez D, Rossi Sebastiano M, Vallaro M, Mileo V, Pizzirani D, Moretti E, et al. Designing soluble PROTACs: strategies and preliminary guidelines. Journal of Medicinal Chemistry. 2022;65(19):12639-49.
8. Saraswat AL, Vartak R, Hegazy R, Patel A, Patel K. Drug delivery challenges and formulation aspects of proteolysis targeting chimera (PROTACs). Drug Discovery Today. 2023;28(1):103387.
9. Pike A, Williamson B, Harlfinger S, Martin S, McGinnity DF. Optimising proteolysis-targeting chimeras (PROTACs) for oral drug delivery: a drug metabolism and pharmacokinetics perspective. Drug Discovery Today. 2020;25(10):1793-800.
10. Goracci L, Desantis J, Valeri A, Castellani B, Eleuteri M, Cruciani G. Understanding the metabolism of proteolysis targeting chimeras (PROTACs): the next step toward pharmaceutical applications. Journal of Medicinal Chemistry. 2020;63(20):11615-38.
11. Dang T, Cao W, Xing L. 1.6 PROTAC Bioanalysis: Challenges and Strategies. Drug Metabolism and Pharmacokinetics: Frontiers, Strategies, and Applications. 2025;2:44.
12. Haid RTU, Reichel A. A mechanistic pharmacodynamic modeling framework for the assessment and optimization of proteolysis targeting chimeras (PROTACs). Pharmaceutics. 2023;15(1):195.
13. Chen Y, Liu F, Pal S, Hu Q. Proteolysis-targeting drug delivery system (ProDDS): integrating targeted protein degradation concepts into formulation design. Chemical Society Reviews. 2024.
14. Nguyen TM, Sreekanth V, Deb A, Kokkonda P, Tiwari PK, Donovan KA, et al. Proteolysis-targeting chimeras with reduced off-targets. Nature chemistry. 2024;16(2):218-28.
15. Jochem M, Schrempf A, Wagner L-M, Segal D, Cisneros J, Ng A, et al. Degradome analysis to identify direct protein substrates of small-molecule degraders. Cell chemical biology. 2025;32(1):192-200. e6.
16. Zheng S, Tan Y, Wang Z, Li C, Zhang Z, Sang X, et al. Accelerated rational PROTAC design via deep learning and molecular simulations. Nature Machine Intelligence. 2022;4(9):739-48.
17. Abouzied AS, Alshammari B, Kari H, Huwaimel B, Alqarni S, Kassab SE. AI-DPAPT: a machine learning framework for predicting PROTAC activity. Molecular Diversity. 2024:1-13.