The FDA Warning Letter issued to DuPont states that the agency considers the microcrystalline cellulose excipient in question, Avicel®, adulterated because its controls for manufacturing, processing, packing, or holding do not conform to GMP, and because the excipient failed to conform to compendial standards for strength, quality, or purity. The action drew industry-wide attention to the role of excipient suppliers in maintaining GMP compliance throughout the drug supply chain.
Since then, a review of FDA enforcement actions in the Redica Systems database shows that five drug GMP Warning Letters have referenced excipients. While none focused primarily on excipients, two cited deficiencies in excipient testing and three noted issues with their use in manufacturing. Broader trends in Form 483 observations are explored below, along with a more detailed look at a recent inspection of a domestic (U.S.) excipient manufacturer, Purac America Inc., where FDA investigators identified significant GMP and data integrity concerns.
Excipient Deficiencies in 483s Since December 2022
Between December 2022 and August 2025, the Redica Systems database records 628 FDA Warning Letters and 2,615 Form 483s. Of these, 90 Form 483s reference excipients, with the term appearing a total of 149 times.
Geographically, the majority of these 483s were issued in the United States (66%), followed by India (14%), China (8%), and the Republic of Korea (4%), with smaller shares across six additional countries.
To better understand the trends, the 149 excipient-related observations have been grouped into categories (see Figure 1). The sections that follow provide examples from each category.
Figure 1. Distribution of Excipient-Related Observations in FDA Form 483s December 2022 - August 2025
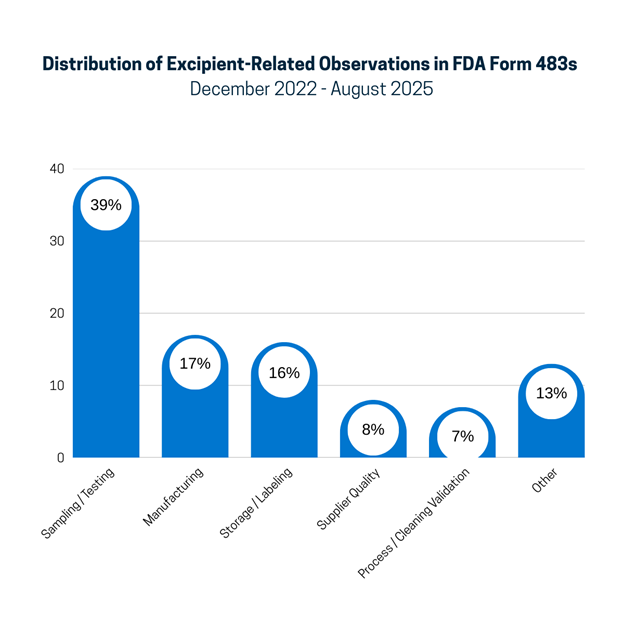
Sampling/Testing:
The largest category, sampling and testing, which represents 39% of the observations, includes a lack of ID testing on receipt, issues with the testing methods used and method validation, and accepting excipients on a COA without testing. For example:
“The firm has not established a procedure for performing at least one specific identity test on excipient components for which the supplier's reports of analysis will be accepted in lieu of performing full testing.”
“Your firm failed to perform method validation of test method protocols for raw materials and excipient (b)(4) testing…”
“The quality unit failed to ensure the active pharmaceutical ingredients and excipients used to manufacture OTC drugs for the US market are tested for identity, purity…”
Manufacturing:
The manufacturing category includes 17% of deficiencies related to the manufacturing of excipients or their use in manufacturing processes. For example:
“Firm is manufacturing active pharmaceutical ingredients (API) and excipients for pharmaceutical use that does not have a controlled batch record…”
“Equipment used in the production, processing, packaging, testing, or storage of an excipient is not maintained in a good state of repair.”
Storage/Labeling
The storage/labeling category represents 16% of excipient findings, for example:
“I observed partial bags of excipients used in manufacturing that were not securely closed, leaving openings that could allow for rodent and insect infestation.”
“The warehouse used for the housing of raw materials, including active pharmaceutical ingredients, excipients, and finished products for distribution has not been temperature mapped to provide a consistent temperature and humidity environment…”
Supplier Quality
Vendor qualification/auditing and quality agreements with suppliers are included in the supplier quality category, responsible for 8% of observations. Examples include:
“Your firm has failed to demonstrate adequate vendor qualification for all your suppliers of API (b)(4) and excipients used in your OTC drug products.”
“Your firm failed to establish procedures and conduct raw materials and/or excipients testing received from your suppliers, nor have you performed supplier qualification to ensure conformity with all written specifications…”
“[SOP] states that a physical audit and quality agreement is required for supplier of active ingredient and supplier of excipients. Your firm does not have a documented quality agreement with your incoming raw material supplier, (b)(4) delineating CGMP responsibilities and has not conducted a physical audit.”
Process/Cleaning Validation
Any mention of excipients as part of process validation or cleaning validation is included in this category, accounting for 7% of the excipient observations. For example:
“Your process validation studies do not take into consideration the variability of the mother mixture concentrate and excipients for each product in each chassis.”
“You describe your finished drug product as a “thin” melt 2- layer tablet with an appropriate ratio of API (Benzocaine) and excipient to each layer. The manufacturing of your two layer orally disintegrating drug tablet consists of mixing the API and excipients in transferring this mixture into a tablet press (WA-EQ- ). However, the process validation for mixing and tableting has no evidence that provides a high degree of assurance that your manufacturing process will result in the desired strength, purity, and quality attributes related to your orally disintegrating drug tablet.”
“No other laboratory testing is performed to ensure cleaning effectiveness and deactivation of hazardous APIs and or excipients in the prevention of cross contamination…”
Other
The “Other” category (13%) primarily consists of the occurrence of the term “excipient” in a way that is not part of a GMP deficiency.
Purac America’s Form 483: Data Integrity and GMP Failures
From July 29 to August 2, 2024, FDA investigators Veronica Fuentes and Steven A. Brettler inspected excipient manufacturer Purac America in Tucker, Georgia. The inspection resulted in a six-page, five-observation Form 483 that documented serious deficiencies across the firm’s Quality, Laboratory, Facilities and Equipment, and Manufacturing systems, including data integrity shortcomings.
Figure 2. Purac America, Inc. Form 483 in the Redica Systems Platform
Quality Unit
The most significant issues centered on the firm’s quality system. Investigators wrote that Purac’s quality unit “failed to develop a quality management system that incorporates the principles of good manufacturing practices (GMP) for excipients.” Examples cited include not following the firm’s deviation and change management procedures and violating fundamental principles of data integrity.
“An original Certificate of Analysis…was removed, destroyed, and replaced with an updated COA template without initiating and completing a deviation, risk assessment, and change control for the updated template. The original record is irretrievable for confirmation and traceability.”
Laboratory
In the laboratory, investigators noted deficiencies primarily with the integrity of documentation, audit trails, and system controls that allow anyone in the lab to make changes to the lab results.
“Electronic records are used, but they do not meet system access limitation, audit trail and systems documentation control requirements to ensure that they are trustworthy, reliable and generally equivalent to paper records.”
For example, one of the FDA investigators wrote on the form 483 that the computer connected to the firm’s equipment used to conduct Inherent Viscosity (IV) testing “was found to have a common username and password for anyone with access to the QC Laboratory to log onto the desktop and access an excel spreadsheet with original, raw data without additional controls to ensure the data within the (b)(4) spreadsheet is secured and tracked for any changes. A QC technician was able to edit raw, original data on the Excel logbook while in my presence.”
These are clear violations of the fundamental principles of computer system security and data integrity.
Testing specifications and expected yields were also called into question.
“The Quality Unit failed to provide a written, scientific justification on why the purification percentage yield is very wide (b)(4) in the manufacture of (b)(4) at your facility where the large, acceptable variability in the percentage yield alone may not be robust to identify a potential issue within the purification operation of manufacturing at the yield reconciliation review and the range is not documented as part of the batch manufacturing record for comparison and review.”
Facilities and Equipment
Equipment and utensils are not adequately cleaned, maintained, and sanitized to prevent contamination, the 483 states. Cleaning validations lacked microbial and chemical residue testing, relying solely on visual inspections. Clean and dirty hold time studies were not performed or documented, and instances of extended periods between cleaning and use were observed. For example,
“We observed an instance that the Grinding and Milling equipment was cleaned on 04/11/2024 and it was used on 04/23/2024 (12 calendar days) without prior additional cleaning. Additionally, we observed an instance that your Grinding and Milling equipment was used on 02/07/2024 and was not cleaned until 04/01/2024 (greater than 30 calendar days).”
Cleanroom certifications by an outside vendor were deemed deficient “due to the certifications not being performed under worst-case conditions for occupancy in the cleanrooms, including but not limited to your ISO classification 6 room.”
Another certification by a third-party vendor was also deemed “deficient due to the lack of non-viable monitoring in the ISO 6 Packaging room for particles of (b)(4) micro-meter particles in size.”
Production
Production practices raised concerns about contamination control. Protective apparel was not consistently worn to prevent drug product contamination. Gowning requirements were not always followed. During the inspection, the FDA investigators
“entered and left the ISO 6 classified room without donning and removing gloves, which is part of the gowning requirements posted as a placard next to the door to enter the room.”
Personnel were observed misusing face masks and failing to follow gowning procedures when entering the ISO 6 classified room. Additionally, the gowning procedure lacks differentiation between ISO 8, ISO 7, and ISO 6 cleanroom requirements.
Key Takeaways from the Purac 483
Although the Form 483 does not indicate the excipients manufactured are considered adulterated, as it did in the Dupont 2022 Warning Letter, serious deficiencies were observed in the Quality, Laboratory, Facilities and Equipment, and Manufacturing systems. Data integrity shortcomings were also uncovered.
The FDA was not the only government agency that found issues at the Georgia plant.
The following month after the FDA inspection, September 2024, the U.S. Environmental Protection Agency (EPA) announced that it had "reached a settlement with Purac America, Inc., doing business as Corbion (Corbion), to resolve alleged violations of federal and state laws governing the handling and storage of hazardous waste at its facility in Tucker, Georgia.”
The company agreed to pay a “$332,000 penalty to resolve the alleged violations and has agreed to perform a Supplemental Environmental Project (SEP) to improve hazardous waste storage practices” at the facility.”